
Member In









Recommended Journals

Ophthalmology and Vision Science

Oral Health and Dentistry

Orthopaedic Surgery and Traumatology

Nutrition and Food Toxicology

Gynaecology and Perinatology

Anaesthesia, Critical Care and Pain Management

Holistic Approaches in Oncotherapy

Current Opinions in Neurological Science

Therapeutic Advances in Cardiology

Multidisciplinary Advances inVeterinary Science

Chronicles of Pharmaceutical Science

Innovative Techniques in Agriculture

Clinical Biotechnology and Microbiology

Pulmonology Research and Respiratory Care

Chronicle of Medicine and Surgery

Archives of Endocrinology and Diabetes Care

Bulletin of Clinical Immunology

Medical Research and Clinical Case Reports
Research Article
Volume 2 Issue 2 - 2018
Development and Evaluation of Floating Pulsatile Drug Delivery System of Meloxicam
1Talla Padmavathi College of Pharmacy, India
2Vaagdevi College of Pharmacy
2Vaagdevi College of Pharmacy
*Corresponding Author: Srilatha Malvey, Talla Padmavathi College of Pharmacy, India.
Received: March 27, 2018; Published: April 04, 2018
Abstract
The main objective of the present research work was to develop chronomodulated, floating-pulsatile drug delivery system to release the drug in distal small intestine part of the GIT in order to achieve chronotherapeutics release of meloxicam without releasing the drug during floating, for the treatment of rheumatoid arthritis, osteoarthritis, spondylitis and to improve the patient compliance. A chronodelivery system, based on biological rhythms, is a state-of-the-art technology for drug delivery to increase safety, efficacy and also improves overall drug performance. The prepared floating-pulsatile tablets of meloxicam consist of different concentrations of a superdisintegrant as a core tablet, containing the active ingredient followed by an erodible membrane of HPMC E5, E15, E50 and topmost covered by buoyant layer consists of HPMC K4M and NaHCO3 (80:20). Developed formulations were evaluated with pre and post compression parameters. On the basis of these evaluation parameters, it was found that optimized immediate release formulation C-3 shows release in 60 min. For pulsatile release core tablet coated with HPMC E 50, P4 shows 7.15 hrs of release, followed by buoyancy layer consists of HPMC K4M and sodium bicarbonate (80:20) showed suitable lag time of 5.15 hrs. Finally, FP4 is considered as an optimized formulation with chronotherapeutic effect.
Keywords: Anti-Inflammatory; Floating Pulsatile; Chronotherapeutics; Rheumatoid arthritis
Introduction
There are many types of arthritis over 100 identified among them the most common are osteoarthritis, rheumatoid arthritis, which make 1% of world population are affected by this chronic diseases, among them women are effected three times more likely to develop rheumatoid arthritis than men, an anti-inflammatory effects of meloxicam are believed to be inhibit the prostaglandin synthetase (cyclooxygenase), leading to the inhibition of prostaglandin synthesis. As prostaglandins sensitize pain receptors, inhibition of their synthesis may be associated with the analgesic and antipyretic effects of meloxicam. Several types of research are going on for the development of new drug delivery system. In conventional therapy, drug is released immediately after medication. So the drug concentration in the plasma is raised and sometimes it is more than the toxic level.
The target of drug discovery is to obtain maximum drug efficacy and minimum side effect. With the advancement of technologies in the pharmaceutical field, drug therapy has changed its path. Controlled release drug delivery systems offer many advantages when compared to immediate release delivery systems, such as nearly constant drug level in the plasma, minimizing the peak-trough fluctuations, avoidance undesirable side effects, reduced dose, reduced frequency of administration, improved patient compliance. Although sustained and controlled release systems have been developed biological systems are not so responsive to these release systems. In addition, sustained and controlled release devices are not applicable in some cases like time-programmed administration of hormones and many drugs which obey chronotherapeutics. Those hormones and drugs can also easily be degraded by metabolic enzymes and resistance may be developed.
The living systems are predictable dynamic resonating systems which require different amounts of drug at expected times within the circadian cycle (Youan, 2004). It is not a surprise that all bodily functions from the single cell to the genome are organized and synchronized in time (Lemmer, 2007). Pulsatile drug delivery system has fulfilled this requirement. Pulsatile drug release is such a system where a drug is released suddenly after well-defined lag time or time gap at desired site within the intestine tract, according to the circadian rhythm of disease states (Busserman., et al. 2003). No drug is released from the device within this lag time is advantageous for the following drugs and therapies.
Many body functions follow the circadian rhythm, i.e., their activity increases or decreases with time. A number of hormones like rennin, aldosterone, and cortisol show daily as well as timely fluctuations in their blood levels. Circadian effects are also observed in case of pH and acid secretion in the stomach, gastric emptying, and gastrointestinal blood transfusion. (Goo., et al. 1987).
The severity of diseases like bronchial asthma, myocardial infarction, angina pectoris, rheumatic disease, ulcer, and hypertension is time dependent (Lemmer., et al. 1999).
To overcome this, novel/conceptual approach termed as “floating pulsatile drug delivery system” was developed. The basic features of this delivery system comprise
To overcome this, novel/conceptual approach termed as “floating pulsatile drug delivery system” was developed. The basic features of this delivery system comprise
- combination of gastro retentive and pulsatile principles,
- an idealistic drug release profile delivering higher amounts of drug at morning time,
- low drug release in stomach suited for the no steroidal anti-inflammatory drug (NSAID)
The sharp increase in asthmatic attacks during early morning hours have been reported (Dethlefsen., et al. 1985) such a condition demands supplement of a drug at the particular time rather than maintaining constant plasma drug level. A drug delivery system administered at bedtime, but releasing drug as a burst after the time of administration (during morning hours) would be ideal in this case. Same is true for preventing heart attacks in the middle of the night and the morning stiffness typical of people suffering from arthritis.
Some drugs (e.g. Salbutamol sulfate) produce biological tolerance and hence the demand for a system that will prevent their continuous presence at the site of action as this tends to reduce their therapeutic effect (Chang., et al. 1999).
Protection from the gastric environment is essential for the drugs that undergo degradation in gastric acidic medium (eg, peptide drugs), irritate the gastric mucosa (NSAIDS) or induce nausea and vomiting. These conditions can be satisfactorily handled by an enteric coating, and in this sense, enteric coating can be considered as a pulsatile drug delivery system.
To achieve localized action at distal organs of GIT such as colon for drugs used in ulcerative colitis (e.g. Sulfasalazine) the drug release needs to be prevented in the upper two-thirds portion of the GIT (Gazzaniga., et al. 1994)
The drugs that undergo extensive first-pass metabolism (ß-blockers) and those that are characterized by idiosyncratic pharmacokinetics or pharmacodynamics resulting in reduced bioavailability, altered drug/metabolite ratios, altered steady-state levels of drug and metabolite, and potential food-drug interactions require delayed release of the drug to the extent possible (Gothoskar., et al. 2004).
All of these conditions demand an efficiently programmed drug delivery system releasing the right amount of drug at the right time. This can be achieved by Pulsatile Drug Delivery Systems. A pulsatile drug delivery system is characterized by a rapid drug release after a predetermined lag time that is an interval of no drug release.
Classification of pulsatile system
;)
Materials & Methods
Meloxicam was kindly gifted by NATCO Pharm, HYD. Perlitol, Crospovidone, Magnesium stearate, Talc, HPMC E5, E15, E50, HPMC K4M, Sodium bicarbonate were obtained from H.D. fine chemicals, Mumbai, India. All other reagents used were of analytical grade.
Preparation of calibration curve of meloxicam
Accurately weighed amount of 100mg drug was transferred to the 100 ml volumetric flask. A small amount of methanol was added to dissolve drug and makeup to 100 ml with methanol to give 1mg/ml solution (Stock 1). Take 10ml solution from the stock 1 into 100 ml volumetric flask and makeup to 100ml with simulated gastric fluid without enzymes (SGF) to give 100μg/ml (Stock 2). Take 10ml from the stock 2 into 100 ml volumetric flask and makeup to 100 ml with SGF (Stock 3). From the stock 3 take 2 ml, 4 ml, 6 ml, 8 ml solutions into the 10ml volumetric flask and makeup to 10 ml with SGF to give 2μg/ml, 4μg/ml, 6μg/ml, 8μg/ml respectively. From the stock 2 take 1 ml, 1.2 ml solutions into 10ml volumetric flask and makeup to 10ml with SGF to give 10μg/ml, 12μg/ml respectively. Prepared samples analyzed by using UV spectrophotometer at the λ max 269 nm.
Accurately weighed amount of 100mg drug was transferred to the 100 ml volumetric flask. A small amount of methanol was added to dissolve drug and makeup to 100 ml with methanol to give 1mg/ml solution (Stock 1). Take 10ml solution from the stock 1 into 100 ml volumetric flask and makeup to 100ml with simulated gastric fluid without enzymes (SGF) to give 100μg/ml (Stock 2). Take 10ml from the stock 2 into 100 ml volumetric flask and makeup to 100 ml with SGF (Stock 3). From the stock 3 take 2 ml, 4 ml, 6 ml, 8 ml solutions into the 10ml volumetric flask and makeup to 10 ml with SGF to give 2μg/ml, 4μg/ml, 6μg/ml, 8μg/ml respectively. From the stock 2 take 1 ml, 1.2 ml solutions into 10ml volumetric flask and makeup to 10ml with SGF to give 10μg/ml, 12μg/ml respectively. Prepared samples analyzed by using UV spectrophotometer at the λ max 269 nm.
Standard calibration curve of Meloxicam
The standard graph of Meloxicam (Table 1) has shown good linearity with R2 values 0.993 and 0.996 in Simulated gastric fluid (without enzyme) (Figure 1) and simulated intestinal fluid (without enzymes) (Figure 2) respectively, which suggests that it obeys the “Beer-Lambert’s law”.
The standard graph of Meloxicam (Table 1) has shown good linearity with R2 values 0.993 and 0.996 in Simulated gastric fluid (without enzyme) (Figure 1) and simulated intestinal fluid (without enzymes) (Figure 2) respectively, which suggests that it obeys the “Beer-Lambert’s law”.
| Concentration (μg/ml) | Absorbance | |
| Simulated gastric fluid | Simulated intestinal fluid | |
| 0 | 0 | 0 |
| 2 | 0.153 | 0.111 |
| 4 | 0.291 | 0.213 |
| 6 | 0.394 | 0.282 |
| 8 | 0.517 | 0.395 |
| 10 | 0.61 | 0.466 |
| 12 | 0.71 | 0.535 |
Table 1: Standard calibration curve of Meloxicam.
;)
Figure 1: Standard graph of meloxicam in Simulate gastric fluid (SGF) without enzymes.
Preparation of core tablets
The composition of the tablets is given in (Table 2). The core tablets containing drug (meloxicam), and superdisintegrant as crospovidone, were prepared by weighing the drug, crospovidone and mixing with spray dried mannitol. Magnesium stearate and talc were added to each blend and further mixed. The resultant blends were tableted to 100 mg using 6 mm flat-faced punches using rotary tablet punching machine (single station tablet compression machine, Cadmach, Ahmedabad, India).
The composition of the tablets is given in (Table 2). The core tablets containing drug (meloxicam), and superdisintegrant as crospovidone, were prepared by weighing the drug, crospovidone and mixing with spray dried mannitol. Magnesium stearate and talc were added to each blend and further mixed. The resultant blends were tableted to 100 mg using 6 mm flat-faced punches using rotary tablet punching machine (single station tablet compression machine, Cadmach, Ahmedabad, India).
| Ingredients(mg)/Formulation | C1 | C2 | C3 |
| Drug | 7.5 | 7.5 | 7.5 |
| Perlitol | 83.5 | 81.5 | 79.5 |
| Crospovidone | 6 | 8 | 10 |
| Magnesium stearate | 1.5 | 1.5 | 1.5 |
| Talc | 1.5 | 1.5 | 1.5 |
| Total | 100 | 100 | 100 |
Table 2: Composition of Core tablets.
Preparation of pulsatile release tablets (PRT)
The core tablets containing meloxicam were compression-coated with 200 mg low-viscosity HPMC E50 in two steps. The first 100-mg coatings were filled into the die, followed by core tablet in the center of the die, and slightly pressed to fix the coatings around and under the core, and then the rest of the coatings were filled and compressed. So PRT dry-coated with 200 mg Methocel E50 was prepared (10mm in diameter, the mean height was 4.45 mm). The composition details are given in the (table 6).
The core tablets containing meloxicam were compression-coated with 200 mg low-viscosity HPMC E50 in two steps. The first 100-mg coatings were filled into the die, followed by core tablet in the center of the die, and slightly pressed to fix the coatings around and under the core, and then the rest of the coatings were filled and compressed. So PRT dry-coated with 200 mg Methocel E50 was prepared (10mm in diameter, the mean height was 4.45 mm). The composition details are given in the (table 6).
Preparation of Floating pulsatile release tablets (FPRT)
FPRT was designed to comprise PRT and a top cover buoyant layer. PRT was taken as the layer for pulsatile release. The buoyant layer included HPMC K4M, which upon contact with gastric fluid formed a gelatinous mass, sufficient for cohesively binding the drug release layer and effervescent component such as sodium bicarbonate, which is fabricated so that upon arrival in the stomach, carbon dioxide is liberated by the acidity of the gastric contents and is entrapped in the jellified hydrocolloid. This produces an upward motion of the dosage form and maintains its buoyancy.
FPRT was designed to comprise PRT and a top cover buoyant layer. PRT was taken as the layer for pulsatile release. The buoyant layer included HPMC K4M, which upon contact with gastric fluid formed a gelatinous mass, sufficient for cohesively binding the drug release layer and effervescent component such as sodium bicarbonate, which is fabricated so that upon arrival in the stomach, carbon dioxide is liberated by the acidity of the gastric contents and is entrapped in the jellified hydrocolloid. This produces an upward motion of the dosage form and maintains its buoyancy.
The buoyant powder of 80% (w/w) HPMC K4M and 20% (w/w) NaHCO3 was passed through a 210-mm sieve to obtain a well-dispersed mixture and followed by the addition of 1% (w/w) magnesium stearate. The 100-mg buoyant powder was filled into the die, followed by PRT in the die, and then compressed as shown in (table 11).
Evaluation of Pre-Compression Blend
Angle of Repose
The angle of repose of granules was determined by the funnel method. The accurately weighed powder mixture was taken in a funnel. The height of the funnel was adjusted in such a manner that the tip of the funnel just touched the apex of the heap of the powder. The powder mixture was allowed to flow through the funnel freely onto the surface. The diameter of the powder cone measured an angle of repose was calculated using the following equation (Raghuram., et al. 2003).
θ = tan-1h/r
Angle of Repose
The angle of repose of granules was determined by the funnel method. The accurately weighed powder mixture was taken in a funnel. The height of the funnel was adjusted in such a manner that the tip of the funnel just touched the apex of the heap of the powder. The powder mixture was allowed to flow through the funnel freely onto the surface. The diameter of the powder cone measured an angle of repose was calculated using the following equation (Raghuram., et al. 2003).
θ = tan-1h/r
Where h and r are the height and radius of the powder cone, θ is the angle of repose. The angle of repose values less than 25, 25-30, 30-40, and more than 40 indicates excellent, good, passable, and poor flow properties respectively.
Determination of Bulk Density and Tapped Density
An accurately weighed quantity of the granules/ powder (W) was carefully poured into the graduated cylinder and volume (V0) was measured. Then the graduated cylinder was closed with the lid and set into the tap density tester (USP). The density apparatus was set to 100 tabs and after that, the volume (Vf) was measured and continued operation till the two consecutive readings were less than 2% (Lachman., et al. 1987).
An accurately weighed quantity of the granules/ powder (W) was carefully poured into the graduated cylinder and volume (V0) was measured. Then the graduated cylinder was closed with the lid and set into the tap density tester (USP). The density apparatus was set to 100 tabs and after that, the volume (Vf) was measured and continued operation till the two consecutive readings were less than 2% (Lachman., et al. 1987).
The bulk density and the tapped density were calculated using the following formulae.
Bulk density = W/V0
Tapped density = W/Vf
Where W= Weight of the powder
V0 = Initial volume
Vf = final volume
Bulk density = W/V0
Tapped density = W/Vf
Where W= Weight of the powder
V0 = Initial volume
Vf = final volume
Compressibility Index (Carr’s Index)
Carr’s index (CI) is an important measure that can be obtained from the bulk and tapped densities. In theory, the less compressible a material the more flowable it is (Lachman., et al. 1987).
Carr’s index (CI) is an important measure that can be obtained from the bulk and tapped densities. In theory, the less compressible a material the more flowable it is (Lachman., et al. 1987).
CI = (TD-BD) x 100/TD
Where TD is the tapped density and BD is the bulk density.
Where TD is the tapped density and BD is the bulk density.
Hausner’s Ratio
It is the ratio of tapped density and bulk density. Hauser found that this ratio was related to interparticle friction and, as such, could be used to predict powder flow properties (Naresh., et al. 1987). Generally, a value less than 1.25 indicates good flow properties, which is equivalent to 20% of Carr’s index.
Hausner ratio = (TD/BD)
It is the ratio of tapped density and bulk density. Hauser found that this ratio was related to interparticle friction and, as such, could be used to predict powder flow properties (Naresh., et al. 1987). Generally, a value less than 1.25 indicates good flow properties, which is equivalent to 20% of Carr’s index.
Hausner ratio = (TD/BD)
Evaluation of Post-Compression
Thickness
Twenty tablets from the representative sample were randomly taken and individual tablet thickness was measured by using digital Vernier Caliper. Average thickness and standard deviation values were calculated. It is expressed in mm
Thickness
Twenty tablets from the representative sample were randomly taken and individual tablet thickness was measured by using digital Vernier Caliper. Average thickness and standard deviation values were calculated. It is expressed in mm
Hardness
Tablet hardness was measured by using Monsanto hardness tester. From each batch, three tablets were measured for the hardness and average of six values was noted along with standard deviations. It is expressed in kg/cm2
Tablet hardness was measured by using Monsanto hardness tester. From each batch, three tablets were measured for the hardness and average of six values was noted along with standard deviations. It is expressed in kg/cm2
Friability Test
From each batch, ten tablets were accurately weighed and placed in the friability test apparatus (Roche friabilator). Apparatus was operated at 25 rpm for 4 minutes and tablets were observed while rotating. The tablets were then taken after 100 rotations, deducted and reweighed. The friability was calculated as the percentage weight loss.
From each batch, ten tablets were accurately weighed and placed in the friability test apparatus (Roche friabilator). Apparatus was operated at 25 rpm for 4 minutes and tablets were observed while rotating. The tablets were then taken after 100 rotations, deducted and reweighed. The friability was calculated as the percentage weight loss.
Note: No tablet should stick to the walls of the apparatus. If so, brush the walls with talcum powder. There should be no capping also.
% friability was calculated as follows
% Friability = (W1–W2) x 100/W1
Where W1 = Initial weight of the 20 tablets.
W2 = Final weight of the 20 tablets after testing.
Friability values below 0.8% are generally acceptable.
% friability was calculated as follows
% Friability = (W1–W2) x 100/W1
Where W1 = Initial weight of the 20 tablets.
W2 = Final weight of the 20 tablets after testing.
Friability values below 0.8% are generally acceptable.
Weight Variation Test
To study weight variation individual weights (WI) of 20 tablets from each formulation were noted using electronic balance. Their average weight (WA) was calculated. Percent weight variation was calculated as follows. Average weights of the tablets along with standard deviation values were calculated.
% weight variation = (WA–WI) x 100/WA
To study weight variation individual weights (WI) of 20 tablets from each formulation were noted using electronic balance. Their average weight (WA) was calculated. Percent weight variation was calculated as follows. Average weights of the tablets along with standard deviation values were calculated.
% weight variation = (WA–WI) x 100/WA
As the total tablet weight was 120 mg, according to IP 1996, out of twenty tablets ± 7.5% variation can be allowed for not more than two tablets. According to USP 2004, ± 10% weight variation can be allowed for not more than two tablets out of twenty tablets.
Drug Content (Assay)
The drug content of the matrix tablets was determined according to in-house standards and it meets the requirements if the amount of the active ingredient in each of the 3 tested tablets lies within the range of 90% to 110% of the standard amount.
The drug content of the matrix tablets was determined according to in-house standards and it meets the requirements if the amount of the active ingredient in each of the 3 tested tablets lies within the range of 90% to 110% of the standard amount.
Three tablets were weighed and taken into a mortar and crushed into fine powder. An accurately weighed portion of the powder equivalent to about 100 mg was transferred to a 100 mL volumetric flask containing 100 mL of methanol. It was shaken by mechanical means for 15min.Then it was filtered through what man filter paper (No. 1) and diluted to 10 mL with simulated intestinal fluid without enzymes and absorbance was measured against blank at 269 nm.
In-Vitro Drug Release Characteristics
For core tablets: The dissolution testing of core tablet was carried out using a USP Type II dissolution apparatus at 37 ± 0.5°C in 900 ml dissolution medium simulated intestinal fluid (without enzymes) speed of 50 rpm (n=3).
For core tablets: The dissolution testing of core tablet was carried out using a USP Type II dissolution apparatus at 37 ± 0.5°C in 900 ml dissolution medium simulated intestinal fluid (without enzymes) speed of 50 rpm (n=3).
For pulsatile release tablets
For Pulsatile tablets two sets dissolution studies were carried out using USP Type I dissolution test apparatus. The volume of dissolution medium (900 ml), stirring speed (100 rpm) and temperature of medium (37 ± 0.2°C) were kept same for all dissolution studies (n = 3).
For Pulsatile tablets two sets dissolution studies were carried out using USP Type I dissolution test apparatus. The volume of dissolution medium (900 ml), stirring speed (100 rpm) and temperature of medium (37 ± 0.2°C) were kept same for all dissolution studies (n = 3).
In one set of dissolution studies, SGF was used as dissolution medium and dissolutions were performed for 5 hrs. The second set of dissolution studies were performed using SGF for a time period equivalent to erosion time which varied for each formulation and then subsequently in a simulated intestinal fluid, without enzymes (SIF) till the complete release of a drug.
For Floating- pulsatile release tablets
For Floating-Pulsatile tablets, two sets dissolution studies were carried out using USP Type I dissolution test apparatus. The volume of dissolution medium (900 ml), stirring speed (100 rpm) and temperature of medium (37 ± 0.2°C) were kept same for all dissolution studies (n = 3). The dissolution studies were performed using SGF for a time period equivalent to erosion time and then subsequently in simulated intestinal fluid, without enzymes (SIF) till the complete release of a drug. At appropriate time intervals, 5 mL of the solution was withdrawn, filtered, and assayed by a UV spectrophotometer at 269 nm, while an equal volume of fresh dissolution medium was added to the apparatus. Dissolution tests were performed in triplicate. The lag time was determined by extrapolation of the upward part of release profile to the time axis.
For Floating-Pulsatile tablets, two sets dissolution studies were carried out using USP Type I dissolution test apparatus. The volume of dissolution medium (900 ml), stirring speed (100 rpm) and temperature of medium (37 ± 0.2°C) were kept same for all dissolution studies (n = 3). The dissolution studies were performed using SGF for a time period equivalent to erosion time and then subsequently in simulated intestinal fluid, without enzymes (SIF) till the complete release of a drug. At appropriate time intervals, 5 mL of the solution was withdrawn, filtered, and assayed by a UV spectrophotometer at 269 nm, while an equal volume of fresh dissolution medium was added to the apparatus. Dissolution tests were performed in triplicate. The lag time was determined by extrapolation of the upward part of release profile to the time axis.
Water uptake and Erosion studies
Water uptake and erosion studies were conducted similarly to the in-vitro dissolution studies. At selected time intervals, an individual tablet was withdrawn using the basket. The basket and the tablet were blotted to remove excess liquid and then weighed on an analytical balance. The wetted tablets were then dried in an oven at 105°C for 3 hrs, cooled in a desiccator and weighed again. This procedure was repeated until constant weight was achieved (final dry weight).
Water uptake and erosion studies were conducted similarly to the in-vitro dissolution studies. At selected time intervals, an individual tablet was withdrawn using the basket. The basket and the tablet were blotted to remove excess liquid and then weighed on an analytical balance. The wetted tablets were then dried in an oven at 105°C for 3 hrs, cooled in a desiccator and weighed again. This procedure was repeated until constant weight was achieved (final dry weight).
The extent of erosion (E) was determined from
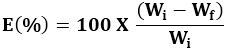
;)
Where Wi and Wf are the initial starting dry weight and final dry weight of the same dried and partially eroded tablet, respectively. The increase in weight (uptake) due to absorbed liquid (A) was calculated at each time point from,
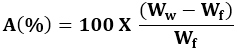 Where Ww is the mass of the wet tablet before drying.
Where Ww is the mass of the wet tablet before drying.
;)
Buoyancy studies
Floating lag time and floating time of FPRT was studied by placing them in 900 mL containers (SGF without enzyme) The floating onset time (time period between placing FPRT in the medium and buoyancy beginning) and floating duration of FPRT were determined by visual observation.
Floating lag time and floating time of FPRT was studied by placing them in 900 mL containers (SGF without enzyme) The floating onset time (time period between placing FPRT in the medium and buoyancy beginning) and floating duration of FPRT were determined by visual observation.
Drug-Excipient compatibility studies
FTIR Studies
FTIR spectra of pure meloxicam, core tablet mixture, and drug-polymer mixture were recorded on Bruker ALPHA model directly placing on probe analyzing for functional groups. Each spectrum was derived from single average scan collected in the region 400-4000 cm-1 at the spectral resolution of 2 cm-2. The obtained spectrums are allowed for peak picking (Naresh., et al. 2016).
FTIR Studies
FTIR spectra of pure meloxicam, core tablet mixture, and drug-polymer mixture were recorded on Bruker ALPHA model directly placing on probe analyzing for functional groups. Each spectrum was derived from single average scan collected in the region 400-4000 cm-1 at the spectral resolution of 2 cm-2. The obtained spectrums are allowed for peak picking (Naresh., et al. 2016).
Stability Studies
The optimized matrix tablets were subjected to stability studies at 25°C ± 2°C/60% ± 5% RH and 40°C ± 2°C/75% ± 5% RH the products were evaluated for their physical characteristics, drug content, and in-vitro drug release profiles over a period of 3 months
The optimized matrix tablets were subjected to stability studies at 25°C ± 2°C/60% ± 5% RH and 40°C ± 2°C/75% ± 5% RH the products were evaluated for their physical characteristics, drug content, and in-vitro drug release profiles over a period of 3 months
Results and Discussion
Characterization of Powder blend
The powder blend of all formulations (C1-C3), (P1-P5), (FP1-FP5), was characterized with respect to the angle of repose, bulk density, tapped density, Carr’s index, and drug content (Table 3). The angle of repose was less than 35° and Carr’s index values were less than 21 for the granules of all the batches indicating good to fair flowability and compressibility. Hausner’s ratio was less than 1.25 for all the batches indicating good flow properties.
The powder blend of all formulations (C1-C3), (P1-P5), (FP1-FP5), was characterized with respect to the angle of repose, bulk density, tapped density, Carr’s index, and drug content (Table 3). The angle of repose was less than 35° and Carr’s index values were less than 21 for the granules of all the batches indicating good to fair flowability and compressibility. Hausner’s ratio was less than 1.25 for all the batches indicating good flow properties.
| Formulation code | Angle of repose | Bulk density | Tapped density | Carr's index | Hausner’s ratio |
| C1 | 26.12 ± 1.13 | 0.311 | 0.365 | 14.79 ± 1.12 | 1.17 |
| C2 | 28.39 ± 1.21 | 0.329 | 0.383 | 14.12 ± 1.2 | 1.16 |
| C3 | 25.79 ± 1.29 | 0.321 | 0.342 | 20.15 ± 1.3 | 1.06 |
| P1 | 30.23 ± 1.23 | 0.332 | 0.345 | 23.33 ± 1.08 | 1.03 |
| P2 | 31.12 ± 1.13 | 0.312 | 0.376 | 24.09 ± 1.05 | 1.20 |
| P3 | 29.13 ± 1.26 | 0.342 | 0.389 | 24.17 ± 1.2 | 1.13 |
| P4 | 32.72 ± 1.23 | 0.319 | 0.406 | 21.43 ± 1.03 | 1.27 |
| P5 | 33.12 ± 1.84 | 0.365 | 0.461 | 20.82 ± 1.04 | 1.26 |
| FP1 | 28.12 ± 1.13 | 0.344 | 0.378 | 23.21 ± 1.21 | 1.09 |
| FP2 | 29.13 ± 1.26 | 0.332 | 0.422 | 21.33 ± 1.3 | 1.27 |
| FP3 | 31.61 ± 1.91 | 0.315 | 0.398 | 20.65 ± 1.03 | 1.26 |
| FP4 | 25.72 ± 1.23 | 0.312 | 0.341 | 22.19 ± 1.29 | 1.09 |
| FP5 | 27.23 ± 1.4 | 0.316 | 0.397 | 20.4 ± 1.01 | 1.25 |
Table 3: Physical evaluation of powder blend.
Physical Evaluation of tablets
The results of the uniformity of weight, hardness, thickness, friability, and drug content of the tablets are given in Table 4. All the tablets of different batches complied with the official requirements of uniformity of weight as their weights varied within the limits. The hardness of the tablets ranged from 2.3 to 6.1 kg/cm2 and the friability values were less than 0.43% indicating that the tablets were compact and hard. The thickness of the tablets ranged from 2.25 to 4.3 mm. All the formulations satisfied the content of the drug as they contained 97.5 to 102.8% of meloxicam and good uniformity in drug content was observed. Thus all the physical attributes of the prepared tablets were found be practically within the limit.
The results of the uniformity of weight, hardness, thickness, friability, and drug content of the tablets are given in Table 4. All the tablets of different batches complied with the official requirements of uniformity of weight as their weights varied within the limits. The hardness of the tablets ranged from 2.3 to 6.1 kg/cm2 and the friability values were less than 0.43% indicating that the tablets were compact and hard. The thickness of the tablets ranged from 2.25 to 4.3 mm. All the formulations satisfied the content of the drug as they contained 97.5 to 102.8% of meloxicam and good uniformity in drug content was observed. Thus all the physical attributes of the prepared tablets were found be practically within the limit.
| Formulation code | Hardness Kg/cm²* | Friability (%) ** | Deviation in weight variation(mg) *** | Thickness(mm)* | Drug content (%)* |
| C1 | 2.3 ± 0.6 | 0.43 ± 0.1 | 99.2 ± 0.7 | 2.25 ± 0.5 | 99.1 ± 0.8 |
| C2 | 2.4 ± 0.8 | 0.38 ± 0.04 | 100.5 ± 1.9 | 2.4 ± 0.7 | 102.8 ± 0.2 |
| C3 | 2.5 ± 0.5 | 0.34 ± 0.07 | 99.8 ± 1.6 | 2.5 ± 0.9 | 98.1 ± 1.2 |
| P1 | 5.3 ± 0.8 | 0.28 ± 0.06 | 351.5 ± 0.9 | 4.2 ± 0.8 | 101.2 ± 0.8 |
| P2 | 5.6 ± 0.2 | 0.40 ± 0.08 | 348.9 ± 2.1 | 4.1 ± 0.9 | 100.8 ± 0.4 |
| P3 | 6.1 ± 0.5 | 0.31 ± 0.04 | 349.4 ± 1.5 | 4.3 ± 0.8 | 97.5 ± 0.9 |
| P4 | 5.5 ± 0.7 | 0.27 ± ± 0.2 | 249.5 ± 1.9 | 3.5 ± 0.5 | 98.6 ± 1.9 |
| P5 | 5.9 ± 0.4 | 0.25 ± 0.06 | 299.9 ± 1.2 | 3.1 ± 0.4 | 100.4 ± 0.9 |
Table 4: Physical Evaluation of prepared tablets.
*All values represent mean ± Standard Deviation (SD), n = 3 ** All values represent mean ± Standard Deviation (SD), n = 6 *** All values represent mean ± Standard Deviation (SD), n = 20
*All values represent mean ± Standard Deviation (SD), n = 3 ** All values represent mean ± Standard Deviation (SD), n = 6 *** All values represent mean ± Standard Deviation (SD), n = 20
In-Vitro Drug Release Studies
For Rapid releasing tablets (RRT)
The results of release studies of formulations C1 to C3 were shown in Table 5, and Figure 3. The release of drug depends on the amount of superdisintegrants which is used in the core tablet formulation. Hence in order to get the rapid release, superdisintegrants was added to the formulation. Superdisintegrants are generally used at a low level in the solid dosage form, typically 1–10% by weight relative to the total weight of the dosage unit. Examples of superdisintegrants are croscarmellose, crospovidone, sodium starch glycolate etc. Crospovidone is a water-insoluble type tablet disintegrant exhibiting high capillary activity and prominent hydration capacity with little tendency to gel formation [Lopez-Solıs., et al. 2001]. The formulation C-1 containing 6% crospovidone disintegrated in 4 min, C-2 (8% crospovidone) in 2 min and C-3 (10% crospovidone) within the fraction of seconds. The drug release was rapid in all formulations. The formulation C-3 showed 86.43 ± 1.87% release in 60 min and found to be suitable to use in pulsatile release tablets
For Rapid releasing tablets (RRT)
The results of release studies of formulations C1 to C3 were shown in Table 5, and Figure 3. The release of drug depends on the amount of superdisintegrants which is used in the core tablet formulation. Hence in order to get the rapid release, superdisintegrants was added to the formulation. Superdisintegrants are generally used at a low level in the solid dosage form, typically 1–10% by weight relative to the total weight of the dosage unit. Examples of superdisintegrants are croscarmellose, crospovidone, sodium starch glycolate etc. Crospovidone is a water-insoluble type tablet disintegrant exhibiting high capillary activity and prominent hydration capacity with little tendency to gel formation [Lopez-Solıs., et al. 2001]. The formulation C-1 containing 6% crospovidone disintegrated in 4 min, C-2 (8% crospovidone) in 2 min and C-3 (10% crospovidone) within the fraction of seconds. The drug release was rapid in all formulations. The formulation C-3 showed 86.43 ± 1.87% release in 60 min and found to be suitable to use in pulsatile release tablets
| Time(min) | C1 | C2 | C3 |
| 0 | 0 | 0 | 0 |
| 10 | 22.14 ± 2.99 | 26.47 ± 1.58 | 42.13 ± 1.8 |
| 20 | 35.69 ± 1.58 | 44.05 ± 1.45 | 53.379 ± 2.1 |
| 30 | 48.28 ± 1.46 | 54.05 ± 2.5 | 62.12 ± 1.49 |
| 45 | 58.56 ± 2.2 | 64.52 ± 2.2 | 75.76 ± 2.3 |
| 60 | 65.67 ± 1.89 | 73.65 ± 1.89 | 86.43 ± 1.87 |
| 90 | 77.78 ± 1.29 | 82.11 ± 2.45 | 94.21 ± 1.68 |
| 120 | 84.41 ± 2.3 | 91.14 ± 2.17 | 100.2 ± 2.6 |
Table 5: In-Vitro Release Data of meloxicam from rapidly releasing tablets (RRT)*.
;)
Figure 2: Standard graph of meloxicam in simulated intestinal fluid (SIF) without enzymes.
;)
Figure 3: In- vitro release profile of rapid release (meloxicam) tablets.
For pulsatile release tablets (PRT)
For in-vitro release studies, the core tablets containing Meloxicam (RRT) were compression-coated with a low-viscosity HPMC (Methocel1 E5, E15, and E50) up to 250 mg (P1, P2, and P3). The in vitro release profiles of Meloxicam from HPMC-coated system were provided in Figure 4. In the in vitro study, the three formulations were designed to have different tablet release profiles by changing weight (150, 200, P4, P5, respectively) of coating layers with HPMC E50. Consequently, differences in tablet release profiles were noted in the in vitro testing as Figure 5.
For in-vitro release studies, the core tablets containing Meloxicam (RRT) were compression-coated with a low-viscosity HPMC (Methocel1 E5, E15, and E50) up to 250 mg (P1, P2, and P3). The in vitro release profiles of Meloxicam from HPMC-coated system were provided in Figure 4. In the in vitro study, the three formulations were designed to have different tablet release profiles by changing weight (150, 200, P4, P5, respectively) of coating layers with HPMC E50. Consequently, differences in tablet release profiles were noted in the in vitro testing as Figure 5.
;)
Figure 4: In-vitro release profiles of Meloxicam from the pulsatile
release tablet (PRT) coated with 250-mg different kinds of HPMC.
;)
Figure 5: In vitro release profiles of meloxicam from the pulsatile
release tablet (PRT) coated with different amount of Methocel E50.
As the coated tablet was placed in the aqueous medium, it was observed that the hydrophilic polymeric layer started erosion, which underwent progressive modification in terms of thickness and consistency. In the second phase of the dissolution procedure, the coating layer gradually starts to erode up to a limiting thickness. After this stage, a rapture of the shell was observed under the pressure applied by the swelling of the core tablet and Meloxicam released. All of this process corresponded to a lag time capable of exhibiting a pulsatile release of the drug. The profiles relevant to the coated tablet showed that a lag phase was followed by the quick delivery of the active principle. The delay duration clearly depended on the kind and amount of hydrophilic polymer which was applied to the core. The lag time in vitro of the tablet coated with 200-mg HPMC E50 was 5.15 ± 0.1 hrs.
| Ingredients | P1 | P2 | P3 | P4 | P5 |
| Drug | 7.5 | 7.5 | 7.5 | 7.5 | 7.5 |
| Perlitol | 83.5 | 83.5 | 83.5 | 83.5 | 83.5 |
| Crospovidone | 6 | 6 | 6 | 6 | 6 |
| Magnesium stearate | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 |
| Talc | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 |
| HPMC E5 | 250 | ||||
| HPMC E15 | 250 | ||||
| HPMC E50 | 250 | 200 | 150 | ||
| Total | 300 | 300 | 300 | 250 | 200 |
Table 6: Composition of PRT.
| Time (hrs) | P1 | P2 | P3 | P4 | P5 |
| 0 | 0 | 0 | 0 | 0 | 0 |
| 1 | 0 | 0 | 0 | 0 | 0 |
| 1.15 | 17.44 ± 2.03 | ||||
| 1.3 | 27.24 ± 2.07 | ||||
| 1.45 | 34.44 ± 1.19 | ||||
| 2 | 52.61 ± 1.96 | 0 | 0 | 0 | 0 |
| 2.3 | 64.28 ± 2.6 | ||||
| 3 | 81.72 ± 2.03 | 0 | 0 | 0 | 0 |
| 3.15 | 4.67 ± 1.6 | 24.93 ± 1.22 | |||
| 3.3 | 18.84 ± 1.4 | 35.31 ± 1.11 | |||
| 3.45 | 37.25 ± 2.4 | 48.86 ± 0.8 | |||
| 4 | 44.85 ± 2.07 | 0 | 0 | 54.91 ± 1.22 | |
| 4.3 | 71.15 ± 1.37 | 61.54 ± 2.07 | |||
| 5 | 84.59 ± 1.66 | 0 | 0 | 83.45 ± 1.63 | |
| 5.15 | 0 | ||||
| 5.3 | 23.35 ± 0.8 | ||||
| 5.45 | 34.88 ± 0.81 | ||||
| 6 | 0 | 46.7 ± 2.6 | |||
| 6.15 | 52.32 ± 2.8 | ||||
| 6.45 | 0 | 62.41 ± 1.6 | |||
| 7.15 | 84.75 ± 1.6 | ||||
| 8 | 0 | ||||
| 8.15 | 7.35 ± .4 | ||||
| 8.3 | 23.92 ± 2.24 | ||||
| 8.45 | 41.22 ± 2.68 | ||||
| 9 | 49.43 ± 1.93 | ||||
| 9.3 | 57.51 ± 2.48 | ||||
| 10 | 83.74 ± 3.2 |
Table 7: In-Vitro Release Data of meloxicam from pulsatile releasing tablets (PRT)*.
| Time (hrs) | FP4 |
| 1 | 0 |
| 2 | 0 |
| 3 | 0 |
| 4 | 0 |
| 5 | 0 |
| 5.15 | 0 |
| 5.3 | 23.35 ± 0.8 |
| 5.45 | 34.88 ± 0.81 |
| 6 | 46.7 ± 2.6 |
| 6.15 | 52.32 ± 2.8 |
| 6.45 | 62.41 ± 1.6 |
| 7.15 | 84.75 ± 1.6 |
Table 8: In-Vitro Release Data of meloxicam from floating pulsatile releasing tablets (FPRT)*.
;)
Figure 6: In-vitro drug release profile of meloxicam floating pulsatile release tablets (FPRT).
;)
Figure 7: % Erosion studies for pulsatile release tablets.
Water uptake and Erosion studies
Since the rate of swelling and erosion is related and may affect the mechanism and kinetics of drug release, the penetration of the dissolution medium and the erosion of the hydrated tablets were determined. Simultaneously with the water uptake study, the percentage erosion of polymer was determined. The percentage water uptake and erosion of all formulations were given in Table 9 and 10. Results showed that higher % water uptake was observed with HPMC E50 with 250 mg weight of the coat than HPMC E15 and HPMC E5. This may be due to the increasing the viscosity of HPMC E50 polymer and also increasing the thickness also affect the % water uptake. % Erosion was also depending on the thickness and viscosity grade of the polymer. When the increasing thickness, viscosity and polymer concentration of the polymer there is delaying polymer erosion.
Since the rate of swelling and erosion is related and may affect the mechanism and kinetics of drug release, the penetration of the dissolution medium and the erosion of the hydrated tablets were determined. Simultaneously with the water uptake study, the percentage erosion of polymer was determined. The percentage water uptake and erosion of all formulations were given in Table 9 and 10. Results showed that higher % water uptake was observed with HPMC E50 with 250 mg weight of the coat than HPMC E15 and HPMC E5. This may be due to the increasing the viscosity of HPMC E50 polymer and also increasing the thickness also affect the % water uptake. % Erosion was also depending on the thickness and viscosity grade of the polymer. When the increasing thickness, viscosity and polymer concentration of the polymer there is delaying polymer erosion.
;)
Figure 8: Morphological changes of pulsatile release tablets during dissolution.
| Time (hrs) | P1 | P2 | P3 | P4 | P5 |
| 0 | 0 | 0 | 0 | 0 | 0 |
| 0.5 | 102 ± 2.23 | 39 ± 2.24 | 15 ± 2.21 | 24 ± 2.7 | 69 ± 2.54 |
| 1 | 16 ± 2.54 | 148 ± 2.34 | 28 ± 2.32 | 75 ± 2.65 | 164 ± 2.34 |
| 2 | 86 ± 2.94 | 52 ± 2.24 | 176 ± 2.54 | 87 ± 2.45 | |
| 3 | 12 ± 2.5 | 93 ± 2.35 | 103 ± 2.45 | 14 ± 2.24 | |
| 4 | 192 ± 2.15 | 79 ± 2.35 | |||
| 5 | 112 ± 2.67 | 16 ± 2.65 | |||
| 6 | 78 ± 2.63 | ||||
| 7 | 42 ± 2.52 | ||||
| 8 | 10 ± 2.57 |
Table 9: % water uptake studies for pulsatile release tablets.
| Time | P1 | P2 | P3 | P4 | P5 |
| 0 | 0 | 0 | 0 | 0 | 0 |
| 0.5 | 76 ± 2.25 | 9 ± 2.34 | 3 ± 2.25 | 4 ± 2.22 | 10 ± 2.14 |
| 1 | 91 ± 2.34 | 24 ± 2.54 | 7 ± 2.35 | 9 ± 2.34 | 32 ± 2.56 |
| 2 | 52 ± 2.54 | 12 ± 2.45 | 25 ± 2.35 | 67 ± 2.76 | |
| 3 | 96 ± 2.9 | 29 ± 2.56 | 41 ± 2.45 | 93 ± 2.78 | |
| 4 | 39 ± 2.67 | 70 ± 2.65 | |||
| 5 | 52 ± 2.87 | 92 ± 2.68 | |||
| 6 | 78 ± 2.98 | ||||
| 7 | 85 ± 2.41 | ||||
| 8 | 95 ± 2.56 |
Table 10: % Erosion studies for pulsatile release tablets.
| Ingredients | Expressed as mg per Tablet | ||||
| FP1 | FP2 | FP3 | FP4 | FP5 | |
| HPMC K4 M | 50 | 70 | 60 | 80 | 90 |
| Sodium bicarbonate | 50 | 30 | 40 | 20 | 10 |
Table 11: The Composition of the Buoyant Layers for Floating Testing.
Buoyancy studies & Composition
The compositions of the buoyant layer of the FPRT for floating testing were shown in (Table 12 and figure 9). All powdered excipients were mixed for 5 min using a mortar and pestle to form a homogenous directly compressible powder mix.
The compositions of the buoyant layer of the FPRT for floating testing were shown in (Table 12 and figure 9). All powdered excipients were mixed for 5 min using a mortar and pestle to form a homogenous directly compressible powder mix.
| Formulation | Floating Onset Time (min) | Floating Duration (h) | Integrity |
| FP1 | <1 | <3 | Broken |
| FP2 | <1 | >12 | Intact |
| FP3 | <1 | >12 | Intact |
| FP4 | <1 | >12 | Intact |
| FP5 | 2-3 | >12 | Intact |
Table 12: Floating of Various Formulation of the Floating-Pulsatile Release Tablet (FPRT).
;)
Figure 9: In-vitro buoyancy studies.
When the system was immersed in a simulated gastric fluid at 37 ± 0.5°C, it sank at once in the solution and formed the swollen tablet with a density much lower than 1 g/ml. The reaction was due to carbon dioxide generated by neutralization in the buoyant layer with the solution. These systems (Tab. FP1, FP2, FP3, FP4, and FP5) were found to float completely within 1 min and remained floating over a period of 12 hrs. The onset time of FP5 floating was about 2–3 min because of very less sodium bicarbonate including the buoyant layers. The duration time of FP1 remaining floating was no more than 3 h because of the too large amount of sodium bicarbonate including the buoyant layers. It is worth mentioning here that sodium bicarbonate, in addition to imparting buoyancy to the novel formulation, provides the initial alkaline microenvironment for polymers to gel. Moreover, the release of CO2 helps to accelerate the hydration of the floating layer, from the above trails optimized composition of HPMC K4M and sodium bicarbonate was selected, (as shown in the table 11), which shows floating onset time is less than 1 min. (as shown in the table 12).
Drug- Excipient compatibility studies
Fourier transform infrared spectroscopy
FT- IR spectrum of pure meloxicam showed characteristic peaks at 1620 cm-1 (C=O stretching), 3292 cm-1 (-NH or –OH) and some prominent bands like 846-567 cm-1 (-CH aromatic ring bending and hetero-aromatics) and 1346-1163 cm-1 (S=O stretching). No drug-polymer interaction was observed in the FT-IR spectra of the powder mixture of optimized formulation since the absorption peaks of the drug still could be detected in the mixture. (as shown in figure 10)
Fourier transform infrared spectroscopy
FT- IR spectrum of pure meloxicam showed characteristic peaks at 1620 cm-1 (C=O stretching), 3292 cm-1 (-NH or –OH) and some prominent bands like 846-567 cm-1 (-CH aromatic ring bending and hetero-aromatics) and 1346-1163 cm-1 (S=O stretching). No drug-polymer interaction was observed in the FT-IR spectra of the powder mixture of optimized formulation since the absorption peaks of the drug still could be detected in the mixture. (as shown in figure 10)
Differential scanning calorimetry
The thermal curve of Meloxicam showed a melting endothermic peak at 268°C (Figure 11). There was no considerable change in the endotherm values of Meloxicam when mixed with excipients (260°C).
The thermal curve of Meloxicam showed a melting endothermic peak at 268°C (Figure 11). There was no considerable change in the endotherm values of Meloxicam when mixed with excipients (260°C).
;)
Figure 10: FTIR spectra of a) Meloxicam alone,
b) Core tablet mixture (C-3) and c) Drug -polymer mixture.
;)
Figure 11: DSC thermo grams of a) Meloxicam alone,
b) drug–polymer mixture and c) core tablet mixture (C3).
Summary
Rapid release tablets of meloxicam were prepared by using different concentrations of super disintegrant (Crospovidone). Pulsatile release tablets were prepared by using different low viscosity grades of HPMC (E5, E15, and E50) and different coat level of the HPMC E50 polymer. The powder blends of all formulations were characterized with respect to the angle of repose, bulk density, tapped density, Carr’s index and Hausner’s ratio. Thus all the physical properties of the prepared powder blend were found be practically within the limit. The prepared tablets were characterized for uniformity of weight, hardness, thickness, friability, and drug content of the tablets.
Thus all the physical properties of the prepared powder blend were found be practically within the limit. The in-vitro dissolution of rapid release tablets was carried out in simulated intestinal fluid without enzymes. The formulation C-3 showed 86.43 ± 1.87% release in 60 min and found to be suitable to use in pulsatile release tablets. The in-vitro dissolution of pulsatile release tablets and floating pulsatile release tablets were carried out using USP type I basket apparatus. The FPRT coated with 200 mg HPMC E50 was shown lag time of 5.15 ± 0.1 h, which was considered suitable lag time for Meloxicam preventing the time-related occurrence of rheumatoid arthritis. The buoyant layer, prepared by 80 mg HPMC K4M, and 20-mg sodium bicarbonate, provided buoyancy to increase the retention of the oral dosage form in the stomach. The PRT coated with HPMC E50 was shown higher % water uptake than the other polymers. It shows delayed erosion compared to HPMC E5 and HPMC E15. This may be due to the higher viscosity of the polymer. Hence FP4 considered as optimized formulation due to its lag time of 5.15 hrs, and drug released was within 7.15 hrs, which is suitable programmed time release.
Conclusion
Prepared floating- pulsatile release tablets containing the floating material, such as HPMC K4M and NaHCO3 (80:20), achieved a satisfactory buoyant force in vitro, whereas the floating lag time was less than 1 min and the floating time was more than 12 hrs. Drug releasing mechanism of FPRT is based on the interaction between hydrophilic polymeric coating and the aqueous gastrointestinal fluids. The in vitro release profiles of meloxicam from FPRT prepared using HPMC E50 (200mg) as retarding polymer are characterized by a predetermined lag phase, the lag time which depends on the kind and amount of the polymeric layer applied on the cores. The developed system offers a simple and novel technique for pulse release of drugs in the stomach or upper part of small intestine.
Acknowledgment
The authors thankful to Talla Padmavathi College of Pharmacy and Vaagdevi College of Pharmacy for providing necessary ingredients to develop this article.
The authors thankful to Talla Padmavathi College of Pharmacy and Vaagdevi College of Pharmacy for providing necessary ingredients to develop this article.
References
- Naresh Kshirasagar., et al. “Design and In Vitro Evaluation of gastro retentive sustained release tablets of ketorolac tromethamine”. Journal of Parma Science 1.2 (2017): 1-24.
- Arkinstall WW. “Review of the North American experience with evening administration of Uniphyl tablets, a once-daily theophylline preparation, in the treatment of nocturnal asthma.” American Journal of Medicine 85.1 (1994): 521-524.
- Avinash R Tekade and Surendra G Gattani. “Development, and evaluation of pulsatile drug delivery system using novel polymer”. Pharmaceutical Development and Technology 14.4 (2009): 380-387.
- Baojian Wu., et al. “Characterization of 5-Fluorouracil Release from Hydroxypropylmethylcellulose Compression-Coated Tablets”. Pharmaceutical Development and Technology 12.2 (2007): 203-210.
- Bin Li., et al. “A novel system for three-pulse drug release based on “tablets in capsule” device”. International Journal of Pharmaceutics 352.1.2 (2008): 159-164.
- Bussemer T., et al. “Evaluation of the swelling, hydration and rupturing properties of the swelling layer of a rupturable pulsatile drug delivery system”. European Journal of Pharmaceutics and Bio pharmaceutics 56.2 (2003): 261-270.
- Bussemer T., et al. “A pulsatile drug delivery system based on Rupturable coated hard gelatin capsules”. Journal of Controlled Release 93.2 (2003): 331-339.
- Chang RK., et al. “Formulation approaches for oral pulsatile drug delivery”. American pharmaceutical review 2.1 (1999): 51-57.
- Cutolo M., et al. “Circadian rhythms in RA”. Annals of the Rheumatic Diseases 62.7 (2003): 593-596.
- Chandu Dabhi., et al. “Predictable pulsatile release of tramadol hydrochloride for chronotherapeutics of arthritis”. Drug Delivery 17.5 (2010): 273-281.
- Dethlefsen U and Repges R. “Ein neues therapieprinzip beinächtlichem asthma”. Medizinische Klinik 44 (1985): 44-47.
- Drayer JI., et al. “Automated ambulatory blood pressure monitoring: a study in age-matched normotensive and hypertensive men”. American Heart Journal 109.6 (1985): 1334-1338.
- Efentakis M., et al. “Design and evaluation of a dry coated drug delivery system with an impermeable cup, swellable top layer and pulsatile release”. International Journal of Pharmaceutics 311.1.2 (2006): 147-156.
- Efentakis M., et al. “Effect of core size and excipients on the lag time and drug release from a pulsatile drug delivery system”. Drug Development and Industrial Pharmacy 37.1 (2011): 113-120.
- Evangelos Karavas., et al. “Application of PVP/HPMC miscible blends with enhanced mucoadhesive properties for adjusting drug release in predictable pulsatile chronotherapeutics”. European Journal of Pharmaceutics and Bio pharmaceutics 64.1 (2006): 115-126.
- Fan TY., et al. “An investigation of pulsatile release tablets with ethylcellulose and Eudragit L as film coating materials and cross-linked polyvinylpyrrolidone in the core tablets”. Journal of Controlled Release 77.3 (2001): 245-251.
- Gandhi BR., et al. “Chronopharmaceutics: As a clinically relevant drug delivery system”. Drug Delivery 18.1 (2011): 1-18.
- Gazzaniga P., et al. “Oral delayed-release system for colonic specific delivery”. International Journal of Pharmaceutics 108.1 (1994): 77-83.
- Goo RH., et al. “Circadian variation in gastric emptying of meals in humans”. Gastroenterology 93.3 (1987): 515-518.
- Hao Z., et al. “Design and Evaluation of a dry coated drug delivery System with Floating–Pulsatile Release”. Journal of Pharmaceutical Sciences 97.1 (2008): 263-273.
- Hiroyuki M., et al. “Formulation Approach for Nicorandil Pulsatile Release Tablet”. Chemical and Pharmaceutical Bulletin 56.4 (2008): 464-467.
- Hoda A Maradny E “Modulation of a Pulsatile Release Drug Delivery System Using Different Swellable/Rupturable Materials”. Drug Delivery 14.8 (2008): 539-546.
- Hao Z., et al. “Design and Gamma-Scintigraphic Evaluation of a Floating and Pulsatile Drug Delivery System Based on an Impermeable Cylinder”. Chemical and Pharmaceutical Bulletin 55.4 (2007): 580-585.
- Janugade B., et al. “Formulation and Evaluation of Press-coated Montelukast Sodium Tablets for Pulsatile Drug Delivery System”. International Journal of Chem Tech Research 1.3 (2009): 24-26.
- Oliver L Freichel and Bernhard C Lippold. “A new oral erosion controlled drug delivery system with a late burst in the release profile”. European Journal of Pharmaceutics and Bio pharmaceutics 50.3 (2000) 345-351.
- Lemmer B. “Chronobiology, drug-delivery and chronotherapeutics”. Advanced Drug Delivery Reviews59 (2007): 825-827.
- Lemmer B. “Cardiovascular chronobiology and chronopharmacology”. Biological Rhythms in Clinical and Laboratory Medicine (1992): 418-427.
- Lemmer B. “Chronopharmacokinetics: implications for drug treatment”. Journal of Pharmacy and Pharmacology 51.8 (1999): 887-890.
- Lida E Kalantzi., et al. “Recent Advances in Oral Pulsatile Drug Delivery”. Recent Patents on Drug Delivery & Formulation 3.1 (2009): 49-63.
- Lopez-Solıs J and Villafuerte-Robles L. “Effect of disintegrants with different hygroscopicity on dissolution of Norfloxacin:Pharmatose DCL 11 tablets”. International Journal of Pharmaceutics 216.1.2 (2001): 127-135.
- Manish Ghimire., et al. “In-vitro/in-vivo correlation of pulsatile drug release from press-coated tablet formulations: A pharmacoscintigraphic study in the beagle dog”. European Journal of Pharmaceutics and Biopharmaceutics 67.2 (2007): 515-523.
- Mandal., et al. “Drug delivery system based on chronobiology—A review”. Journal of Controlled Release 147.3 (2010): 314-325.
- Martin RJ and Banks-Schlegel S. “Chronobiology of asthma”. American Journal of Respiratory and Critical Care Medicine 158.3 (1998): 1002-1007.
- Michael H Smolensky and Nicholas A Peppas b. “Chronobiology, drug delivery, and chronotherapeutics”. Advanced Drug Delivery Reviews 59.9.10 (2007): 828-851.
- Muller JE., et al. “Circadian variation and triggers of onset of acute cardiovascular disease”. Circulation 79.4 (1989): 733-743.
- Pallab Roy and Aliasgar Shahiwala. “Multiparticulate formulation approach to pulsatile drug delivery: Current perspectives”. Journal of Controlled Release 134.2 (2009): 74-80.
- Naresh Kshirasagar., et al. “Formulation and evaluation of orodispersible tablets of naratriptan using sublimation technique”. Indo American journal of pharmaceutical sciences 3.1 (2016): 22-30.
- Ramesh D Parmar., et al. “Pulsatile Drug Delivery Systems: An Overview”. International Journal of Pharmaceutical Sciences and Nanotechnology 2.3 (2009).
- Roland B., et al. “Pulsatile Drug-Delivery system”. Critical Reviews in Therapeutic Drug Carrier System 18.5 (2001): 433-458.
- Shan-Yang Lin., et al. “Hydrophilic Excipients Modulate the Time Lag of Time-Controlled Disintegrating Press-coated Tablets”. AAPS Pharm SciTech 5.4 (2004): 25-29.
- Sher P., et al. “Low density porous carrier based conceptual drug delivery system”. Microporous and Mesoporous Materials 102.1.3 (2007): 290-298.
- Swathi J., et al. “Development of Pulsatile release tablets of Atenolol with swelling and rupturable layers”. International Journal of Applied Pharmaceutics 2.3 (2010): 3140.
- Tofler GH., et al. “Concurrent morning increase in platelet agreeability and the risk of myocardial infarction and sudden cardiac death”. Journal of Medicinal Chemistry 16.24 (1987): 1514-1518.
- Srilatha Malvey., et al. “Formulation and evaluation of acyclovir orodispersible tablets using sublimation method”. Journal of General Practice 3.4 (2015): 1-5.
- Umesh Mukhija., et al. “Time and pH-Dependent Pulsatile Delivery of Meloxicam Solid Dispersions for Rheumatoid Arthritis”. International Journal of Parma Recent Research 2.2 (2010).
- Usha Yogendra Nayak., et al. “Chronotherapeutic drug delivery for early morning surge in blood pressure: A programmable delivery system”. Journal of Controlled Release 136.2 (2009): 125-131.
- Ying Zhu and Liangyuan Zheng. “Development and Mathematical Simulation of Theophylline Pulsatile Release Tablets”. Drug Development and Industrial Pharmacy 31.10 (2005): 1009-1017.
- Youan BC. “Chronopharmaceutics: gimmick or clinically relevant approach to drug delivery?” Journal of Controlled Release 98.3 (2004): 337-353.
- Yao L., et al. “Design and Evaluation of pH-Independent Pulsatile Release Pellets Containing Isosorbide-5-mononitrate”. Chemical and Pharmaceutical Bulletin 57.1 (2009): 55-60.
- Naresh Kshirasagar., et al. “formulation and evaluation of naratriptan orodispersible tablets using super disintergrants by direct compression method”. International journal of pharmaceutical research scholar 2.2 (2013): 268-278.
- Naresh Kshirasagar., et al. “Design and Evaluation of controlled release chitosan microspheres of aceclofenac”. Journal of Medical and Health Research 1.2 (2017): 1-13.
- Zaheeda Khan., et al. “Drug delivery technologies for chronotherapeutic applications”. Pharmaceutical Development and Technology 14.6 (2009): 602-612.
Citation:
Srilatha Malvey and Naresh Kshirasagar. “Development and Evaluation of Floating Pulsatile Drug Delivery System of
Meloxicam”. Chronicles of Pharmaceutical Science 2.2 (2018): 474-492.
Copyright: © 2018 Srilatha Malvey and Naresh Kshirasagar. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
 Scientia Ricerca is licensed and content of this site is available under a Creative Commons Attribution 4.0 International License.
Scientia Ricerca is licensed and content of this site is available under a Creative Commons Attribution 4.0 International License.